Informationen zur EU-Medizinprodukteverordnung MDR und deren Umsetzung bei der Fresenius Kabi Deutschland GmbH
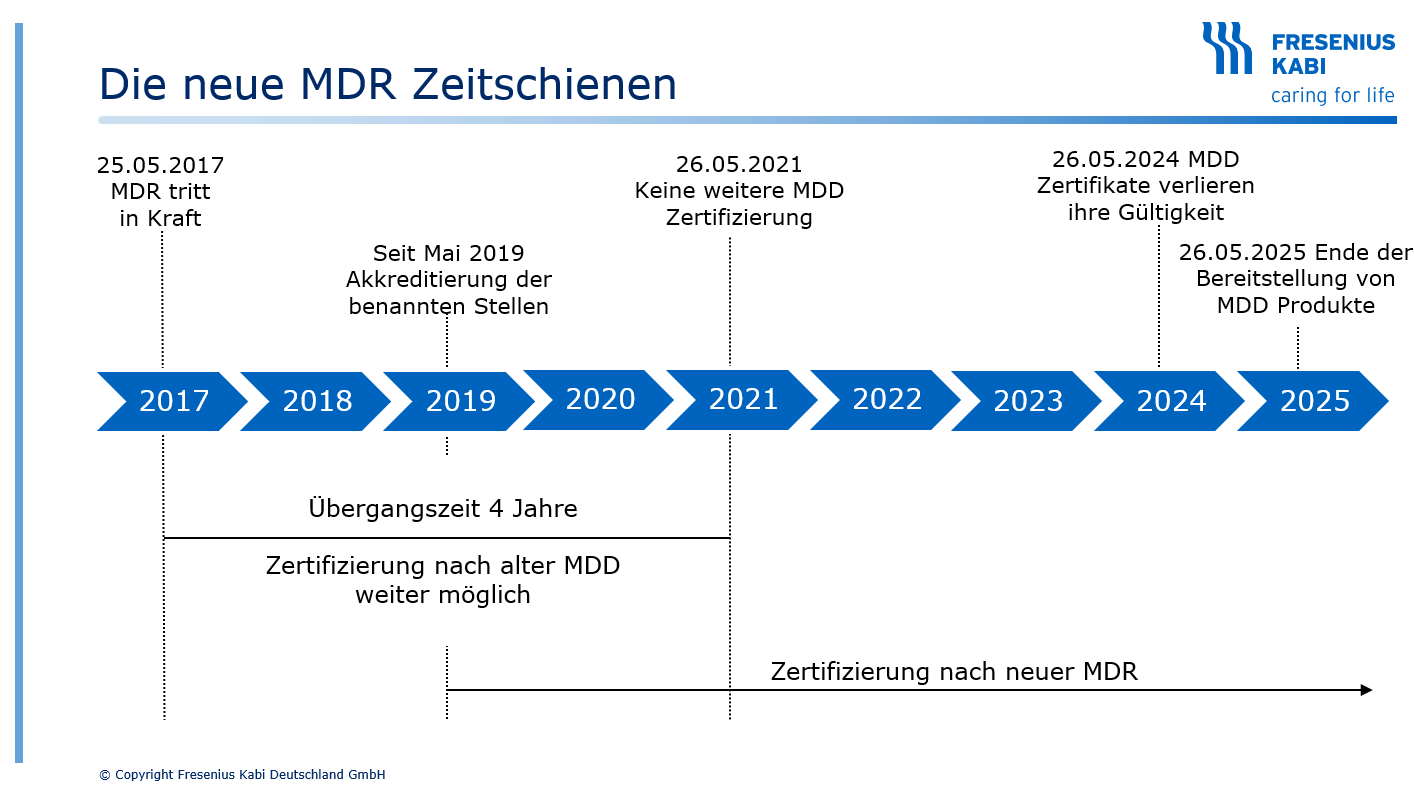
Im Mai 2021 endete die Übergangsfrist für die neue EU-Medizinprodukteverordnung mit vielen neuen Regelungen und Herausforderungen für alle Beteiligten. Fresenius Kabi hat auf dieser Internetseite alle Informationen rund um die MDR für Sie zusammengestellt.
Link zur Datenbank zum Download aller MDR relevanten Dokumente der Fresenius Kabi Produkte
Elektronische Gebrauchsanweisung (eIFU)
Fresenius Kabi stellt Ihnen für für die Medizinprodukte online über diese neue Internetseite elektronische Dokumente zur Verfügung. Somit erfüllen wir die Anforderungen aus Kapitel I, III Artikel 23.1. der MDR (Verordnung EU 2017/745).
Zusätzlich zur elektronischen Gebrauchsanweisung (eIFU) unserer Medizinprodukte stellen wir Ihnen auf dieser Webseite auch noch folgende Informationen zur Verfügung:
- Zusammenfassung der Safety and Clinical Performance (SSCP), solange die EUDAMED noch nicht verfügbar ist
- ISO 13485- und 9001-Zertifikate
- Konformitätserklärungen (DoC)
- Qualitätszertifikate (CoQ)
Diese MDR-relevanten Dokumente finden Sie direkt über folgenden Link:
https://ifu.fresenius-kabi.com/
Auf unserer eIFU-Website angekommen werden Ihnen folgende Informationen angezeigt:

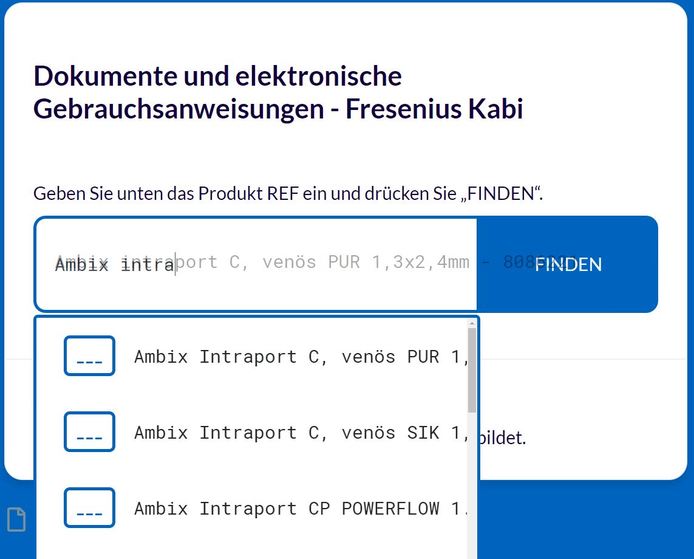
Sie können über die Eingabe einer Artikelnummer oder den Namen des Produkts suchen:

Nachdem Sie den richtigen Artikel gefunden haben, wird z.B. das folgende Fenster angezeigt:
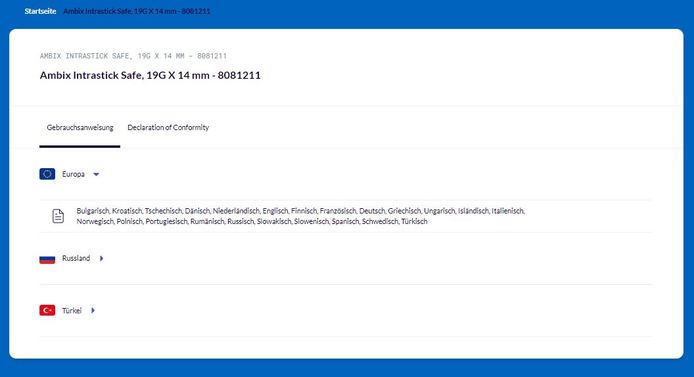
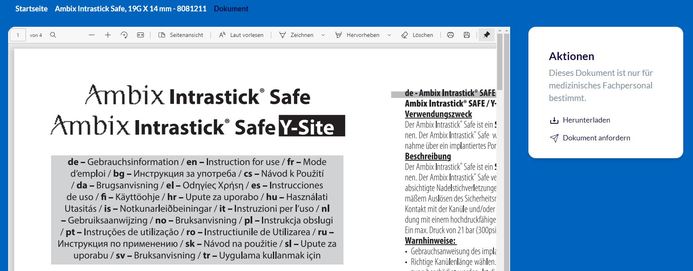
Sie können nun in der Zeile unterhalb des Artikelnamens zwischen den einzelnen Dokumenten auswählen.
Über die Schaltfläche "..." können Sie die Druckversion anfordern oder die Historie der Gebrauchsanweisung einsehen. Über die blaue Schaltfläche "Ansicht" können Sie die aktuell gültige Gebrauchsanweisung lesen und herunterladen.
Für MDR-relevante Dokumente zu unseren sanabelle-Produkten wenden Sie sich direkt an die Kontaktdaten auf folgender Seite:
Informationen zur MDR